Main Body
17. Ion Channels
pore-forming membrane proteins that allow ions to pass through a channel pore
- Ligan-Gated Ion Channels (aka iontropic)
- Ion channels that open in response to specific ligand molecule(s) binding to the receptor protein
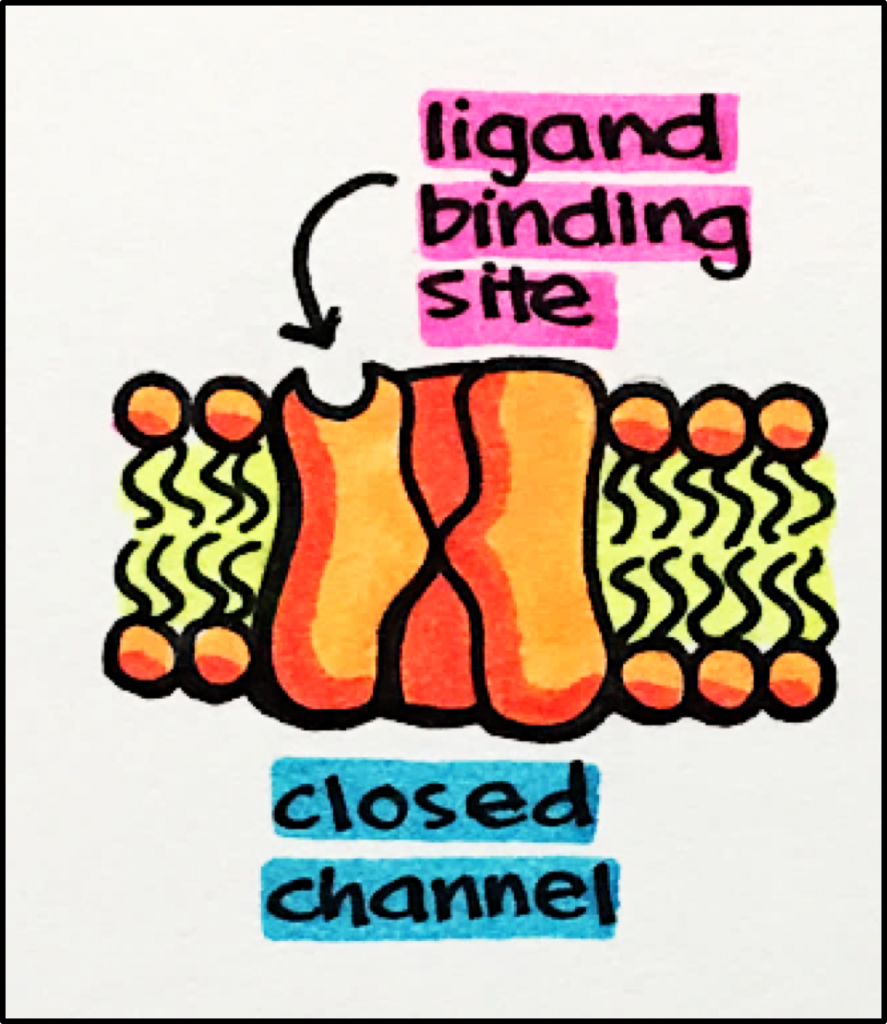
- Ion channels that open in response to specific ligand molecule(s) binding to the receptor protein
- Voltage-Gated Ion Channels
- Ion channels that open and close in response to changes in membrane potential
- Ion channels that open and close in response to changes in membrane potential


Ligand-Gated Ion Channels
Ligand-Gated Ion Channels – allow ions to flow into or out of the cell in response to the binding of a chemical messenger to their respective receptors.
- Acetylcholine Receptors (nicotinic)
- GABAA Receptors
- Glutamate Receptors (NMDAA, AMPA)
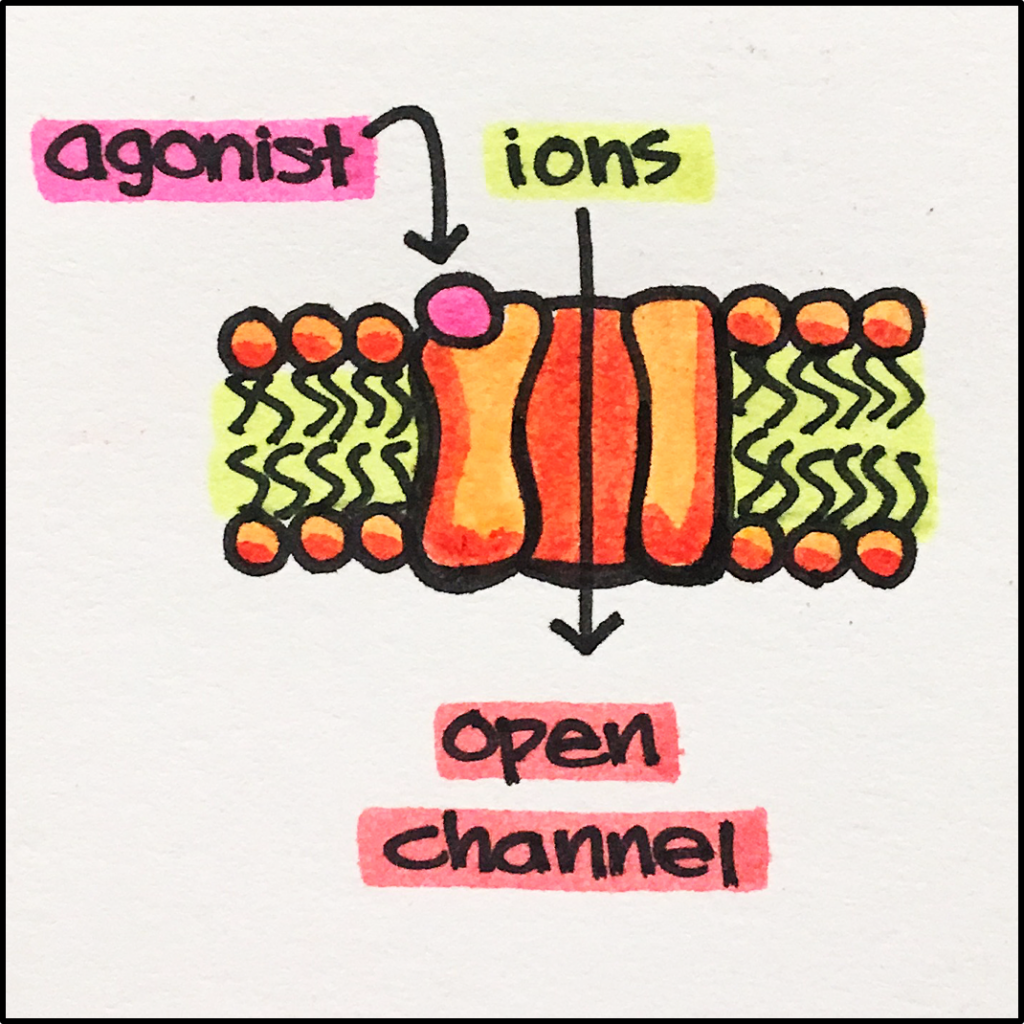
Receptor Stimulation – Ligand binds to the receptor –>> causes receptor conformational change –>> channel opens allowing SPECIFIC ions to pass through.
IONOTROPIC vs METABOTROPIC Receptors
| Ionotropic Receptor | Metabotropic Receptor | |
| Receptor Type | ligand-gated transmembrane protein | ligand-gated transmembrane protein |
| Receptor Structure | ion channel | no ion channel |
| Time to Action | fast acting | slow acting |
| Result of Ligand Binding | ligand directly opens ion channel | ligand activates G-protein leading to a cascade of events that may eventually result in opening an ion channel |
 |
 |
ACETYLCHOLINE RECEPTORS
Acetylcholine Receptors – There are two families of acetylcholine receptors; muscarinic and nicotinic. Both families are found pre-synaptically (ndurons that send neurotransmitter signals) as well as post-synpatically (neurons that receive neurotransmitter signals). They are widely distributed throughout the central nervous system (CNS) and peripheral organs. Certain neurodegenerative diseases, especially Alzheimer’s dementia, are associated with reduction in the number of cholinergic neurons (neurons that synthesize and release acetylcholine).
Nicotinic Acetylcholine Receptors [nAChR] – these channels are pentamers composed of multiple subunits, these channels are directly coupled to cation channels and mediate FAST EXCITATORY synaptic transmission at the NEUROMUSCULAR junction, autonomic ganglia and various sites in the CNS.
- nAChR has 5 subunits and requires two molecules of acetylcholine [ACh] to bind to open the channel (see below).

Muscarinic Acetylcholine Receptors [mAChRs] – mAChRs are G protein coupled receptors [GPCRs] and, therefore, will not be discussed in detail in this section.
nAChR Synapse – nAChRs are found in neuromuscular and ganglionic synapses. Reminder – a synapse is the gap through which electrical or chemical signals are transmitted from a nerve to a muscle or another nerve. Presynaptic nAChRs facilitate ACh release during synaptic activity. Excess neurotransmitter is recycled and broken down by an enzyme called acetylcholinesterase [AChE], rendering ACh inactive as a neurotransmitter.

ACh (seen in the synaptic cleft above) is an important drug target. By inhibiting AChE, ACh is not broken down, the amount of ACh in the synaptic cleft increases, allowing for increased stimulation of synaptic receptors by ACh.
The autonomic system is responsible for controlling those functions we do not consciously control such as breathing, food digestion, heart rate, etc. The autonomic nervous system can be divided into two branches: the sympathetic nervous system [SNS] and the parasympathetic nervous system [PNS]. Nicotinic acetylcholine receptors play an important role in both the SNS as well as the PNS.
Below – In this image, the nicotinic acetylcholine receptor [nAChR] functions to regulate the release of the neurotransmitters [NT] norepinephrine [NE]. Two acetylcholine [ACh] molecules bind the nAChR resulting in the release of NE from the postganglionic fiber. NE will bind to alpha and beta receptors [GPCRs] located on peripheral tissues producing a number of effects associated with a “fight or flight” response such as increased heart rate and contractility, and bronchiole dilation.

Below – In this image, the nAChR also functions to regulate the release of the NT Ach from the postganglionic fiber. ACh goes on to stimulate muscarinic acetylcholine receptors [mAChR; GPCR] located on peripheral tissues producing a number of effects associated with “resting and digesting” such as reduced heart rate and contractility and increased intestinal activity.

GABA (gamma aminobutyric acid) RECEPTORS
GABAA Receptors – these receptors are pentamers (i.e. five subunits) that include a GABA binding site, a channel through which ions (chloride) can travel, as well as a variety of modulatory sites.
GABA is an endogenous agonist synthesized from glutamate
GABA is the main INHIBITORY transmitter in the brain. It acts on both GABAA receptors (GPCRs).
GABAA receptors are ionotropic receptors that allow for chloride ions to flow into a cell upon agonist (GABA) binding. Chloride ions moving into a cell typically decrease second messenger signaling and produce inhibitory effects.
Pharmacological modulators of GABAA receptors include alcohol, barbiturates, benzodiazepines, and neurosteroids (steroids that alter neuronal activity).
Example – Endogenous neurosteroids are synthesized in the brain, adrenals and gonads. These neurosteroids are metabolites of steroids such as progesterone and testosterone; some are positive modulators (agonists) and others are negative modulators (antagonists) of GABAA receptors.
GABAA receptors provide one of the best examples of the application of allosteric modulators. For example, benzodiazepines (e.g., valium) bind to a receptor subunit different from (i.e., allosteric) that where GABA binds. This binding results in altering the conformation of the GABA binding site to a state that has higher affinity for GABA (positive allosteric modulation). This results in potentiating the CNS suppression induced by GABA and explains the effectiveness of benzodiazepines in the treatment of anxiety.

GABAA Antagonists – Decrease central nervous system activity, sometimes used with anesthesia in an inpatient setting. Sedatives, hypnotics, muscle relaxants and anti-convulsants.
GLUTAMATE RECEPTORS
Glutamate Receptors – composed of homo- or hetero-tetramers (four subunits). Found in the CNS on presynaptic neurons that control the release of neurotransmitters. Some glutamate receptors are couple to G proteins (metabotropic) while others are coupled to ion channels (ionotropic).
Types of ionotropic Glutamate Receptors
- AMPA Receptors – Expressed on astrocytes as well on neurons and mediate fast excitatory synaptic transmission.
- Kainate Receptors – Expressed on presynaptic neurons as well as nerve terminals. No known drug targets currently available for this particular receptor.
- NMDA Receptors – Expressed on presynaptic and postsynaptic neurons as well as nerve terminals.
NMDA RECEPTORS
NMDA [N-methyl-D-aspartate] receptors are glutamate-gated cation channels. Once activated, these receptors are highly permeable to sodium and calcium. As mentioned above, NMDA receptors are expressed on presynaptic and postsynaptic neurons as well as nerve terminals. It is at these nerve terminals that glutamate acts on NMDA receptors to alter neurotransmitter release; mainly enhancing it. Furthermore, postsynaptic receptors contribute a slow component to the excitatory synaptic potential.
NMDA receptors have a number of important roles such as in the development of the CNS, rhythmic breathing as well as learning and memory.
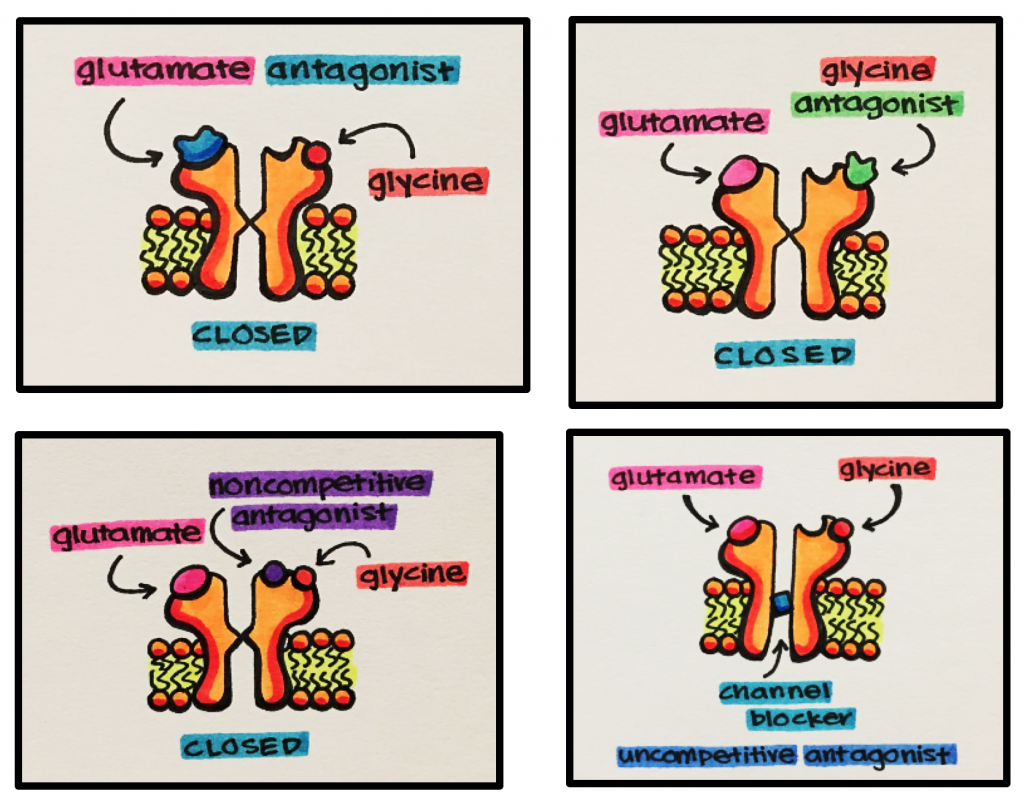
Agonists – NMDA receptors need both glutamate and glycine to bind to the receptor in order to produce a biological effect.
Actions – increased NA+ and Ca2+ influx
Structure – each subunit of the NMDA Receptor has an extracellular N-terminus with 3 transmembrane domains, an intramembrane reentrant “p-loop” between the 1st and 3rd transmemrane domains and an intracellular C-terminus.
- Active Site – site at which agonist (glutamate) binds to stimulate a biological effect or response
- Glycine Binding Site – a specific modulatory site at which glycine binds to the receptor.
- Glycine is a co-agonist. This means that both glutamate and glycine must simultaneously bind and stimulate the receptor in order to produce a physiological effect.
- Allosteric Site – secondary site at which drugs bind to modulate the binding of the agonist to the receptor primary binding site to either enhance (positive allosterism) or reduce (negative allosterism) its effects.
- Channel-blocking Site – site at which drugs bind to block the movement of Na+ and CA2+ ions into the cell
NMDA Antagonists –
- Competitive NMDA Antagonists – block glutamate from binding to the active site
- Glycine Antagonists – bind to the glycine site to block glycine binding, which is necessary for receptor activation
- Noncompetitive Antagonists – inhibit glutamate binding indirectly by binding at negative allosteric sites
- Uncompetitive Antagonists – bind inside the channel pore to block the ion channel and prevent ion movement into and out of a cell
Examples – drugs and their indications
- Anesthetics – dizoclipine (MK801 ®), ketamine (Ketaject ®), phencyclidine
- Ketamine is an anesthetic adjunct. It is indicated for surgical or diagnostic procedures; ideally short procedures. Ketamine is a noncompetitive NMDA receptor antagonist, which means this drug acts at an allosteric site (site other than ligand-binding site) to inhibit receptor function.
- Anti-tussives (cough suppressants) – dextromethorphan/dextrophan is a noncompetitive NMDA receptor antagonist. It is the main active ingredient in Robitussin ®, a widely used cough suppressant
- Alzheimer’s Disease – memantine (Namenda ®)
- Neurodegenerative disorders such as Alzheimer’s disease are caused by abnormally high neuronal excitation (excitotoxicity). Meamantine is a noncompetitive NMDA receptor antagonist that binds to the NMDA receptor cation channel to prevent Ca2+ influx. It is therefore indicated for Alzheimer’s disease.
- Anesthetics – dizoclipine (MK801 ®), ketamine (Ketaject ®), phencyclidine

